 Log In
Log In

























Inventory Management Template
Track current inventory by managing shipments and orders.

Leverage Knack AI to expedite your new app with just a few sentences and save hours of time.

Safeguard data, control access, and ensure robust system reliability for peace of mind.

Organize, import, and manipulate data efficiently to make informed decisions and drive success.

Integrate data and workflows to enhance productivity and expand functionality effortlessly.

Analyze data, create charts, track performance, and make data-driven decisions with ease.

Control permissions, grant or restrict access, and ensure data security effortlessly.

Sell products, manage inventory, and process payments smoothly for your e-commerce business.
Design your app with the best experience for your users and appearance for your brand.

Automate data handling, trigger notifications, and simplify actions for efficient operations.
Expedite your app build with a pre-built template.

Discover valuable insights and tips.
Read Knack success stories.

Download and use a spreadsheet template.

Watch testimonials, tutorials, & expert reviews.

See why Knack is the best option.

Find an expert to build or expand your app.

Learn about how to set up and expand your app.

Become a certified Knack Expert.

Find comprehensive guides and documentation.

Discuss your build with other Knack builders.

Increase your revenue with Knack.
Get hired to build and expand apps.








You can leverage AI to expedite your app build. With up to 400 characters, you can lay the foundation for your entire app.
Choose one of dozens of template apps to customize to your needs.
Craft your app meticulously with Knack for unparalleled customization and control.
Simple tools to build customized apps.
Check out our Interactive Product Tour below to get to know how Knack can work for you, then start building for free.
Your employees already have key industry and process expertise required to iterate faster, let them build and improve the application to their unique requirements.
Build data-rich applications such as SaaS apps, client portals, and internal tools for business processes or online member communities.

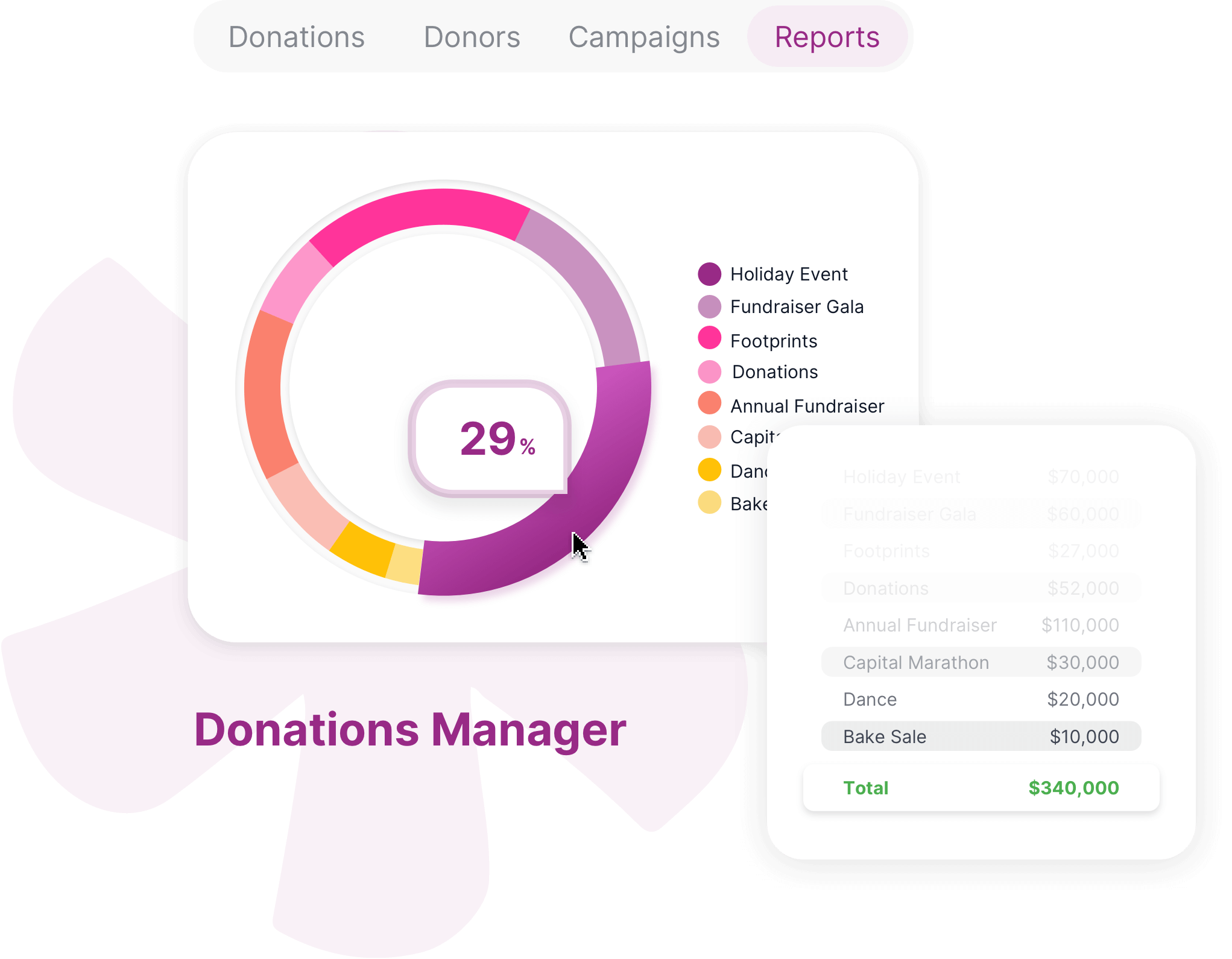
Foster better collaboration by bringing structure to your unstructured processes and tasks with a no code database for project management and more.
Make daily work more impactful by curating and intelligently augmenting your datasets for high-impact applications.
Scale your app-building based on value to the business, not by the number of users. Knack helps you build apps at a fraction of the cost of a traditional pay-per-seat model with an ROI and payback of 6 months or less. Deploy instantly and accelerate new builder adoption with live help from our in-house No Code expert builders.

View the top 10 templates to link to in this view:

Integrate your calendars, cloud storage, email, and other independent tools and sync across your organization.


















“Knack is instrumental in the daily running of the business. It literally runs every facet of our company, from front to back – top to bottom. In the beginning, it probably took me three or four days to build out a new section. Now I can build something new in under an hour”

“We don’t need a full support team or stack of engineers. For some issues, if someone comes to us and has a problem, Knack takes care of it, and that’s it. That’s a huge, huge benefit.”

“The speed of development with Knack was really critical for what we needed, With other programs, even if the features were there, it took more work, you could accomplish the same thing with fewer clicks.”
















